ライゲーション: 原理、プロトコール、トラブル
UBC/experiments/dna/ligation
このページの最終更新日: 2026/04/09広告
ライゲーションの原理と概要
ライゲーション ligation は、DNA リガーゼ ligase という 酵素 を使って DNA 鎖を結合させる反応のことで、主に
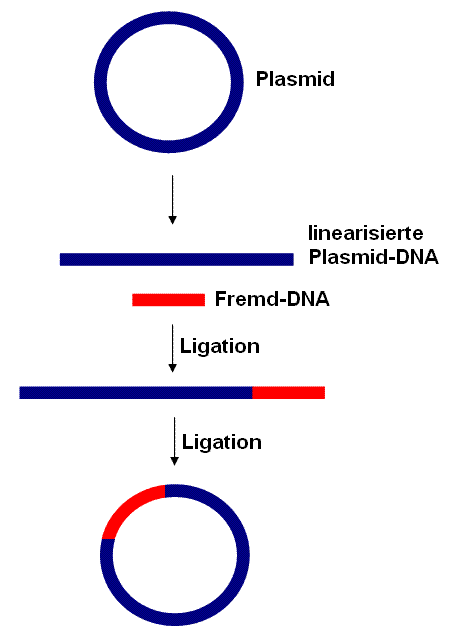 |
 |
DNA のリン酸化
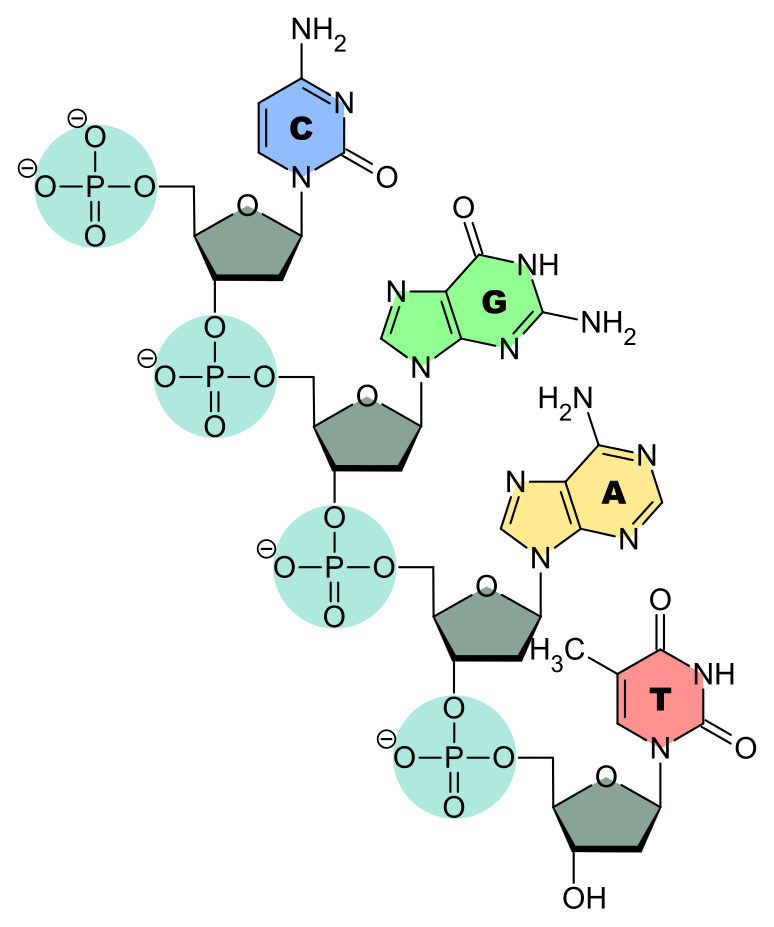
DNA は、図のようにデオキシリボースと塩基がリン酸基を介して繋がった構造をもっている。
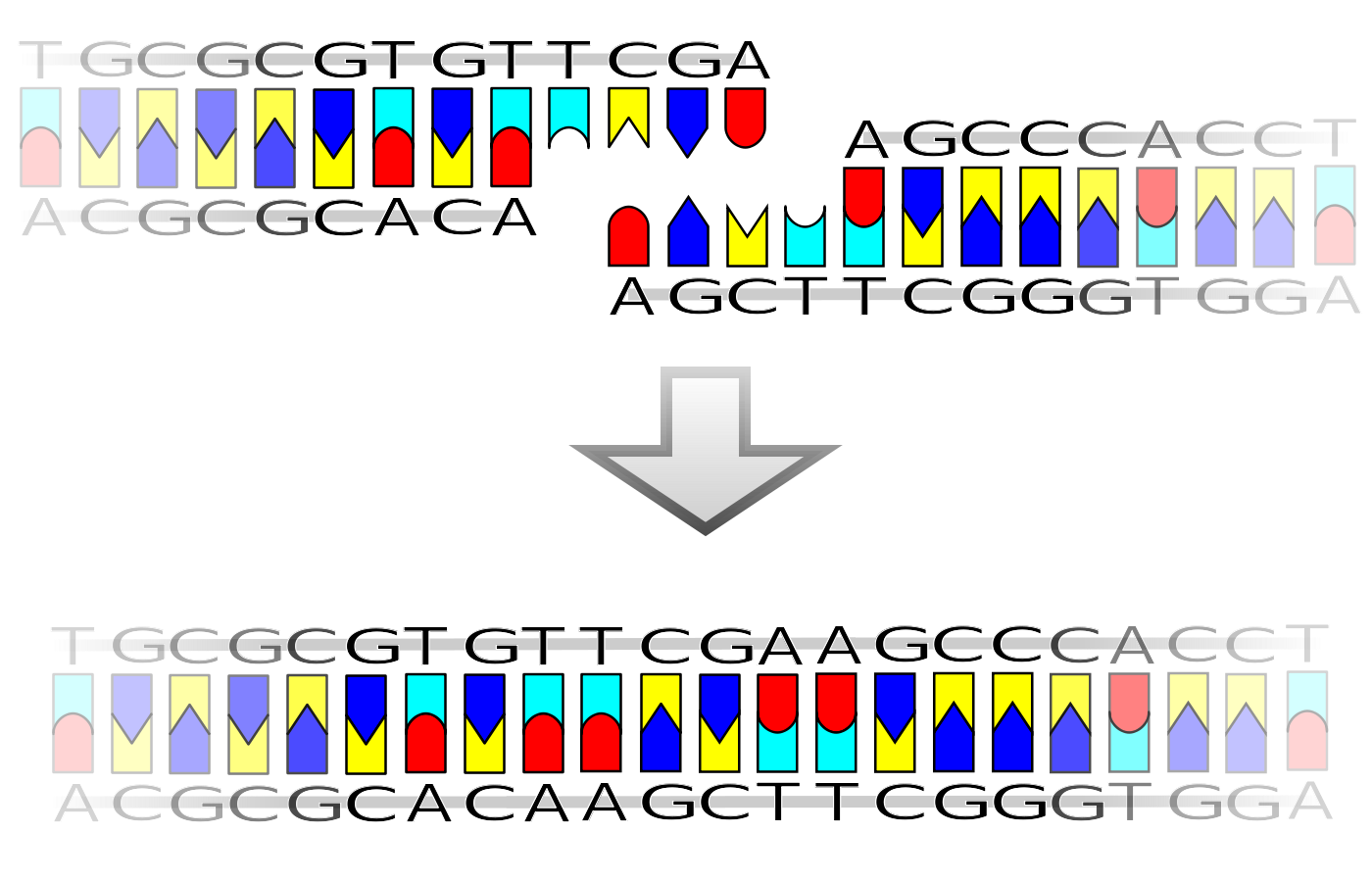
したがって、5' 側にリン酸基がないと、次の塩基と結合できないことがわかるだろう。ライゲーションというのは、5' 側にリン酸基がついた DNA 断片の 5' と、3' 側に -OH 基をもつ DNA 断片の 3' 側を繋げる反応である。
PCR 産物をベクターにライゲーションする場合、PCR 産物は通常はリン酸化されておらず、ベクターにリン酸基がついている。T クローニングの場合にも、既存のベクターを制限酵素で切ったときにもそうである。したがって、意識してリン酸基を導入したりすることはあまりない。
Inverse PCR で PCR 産物を用いたセルフライゲーションを行う際には、それをリン酸化する必要がある。詳細はリンク先を参照のこと。
プロトコール
ライゲーションのプロトコールは使う試薬によって異なるが、ほとんどの場合極めて単純で、試薬を混ぜてインキュベートするだけである。
ベクターとインサートのモル比
経験上、ライゲーション効率を上げるために大切なのは
NEBioCalculator で、ベクターとインサートのサイズから最適な混合量を計算することができる。
ライゲーション反応液の調製
NEB の標準プロトコール。
| 試薬 | 使用量 (20 µL の反応系) |
|---|---|
| 10 x T4 DNA ligase buffer | 2 µL |
| Vector DNA (4 kb) | 50 ng (0.020 pmol) |
| Insert DNA (1 kb) | 37.5 ng (0.060 pmol) |
| Nuclease free water | to 20 µL |
| T4 DNA ligase | 1 µL |
- 上記試薬を混合する。T4 DNA ligase は最後に加える。
- 付着末端 cohesive (sticky) end ライゲーションでは、16℃ O/N または室温で 10 分。
- 平滑末端 blunt end ライゲーションでは、16℃ O/N または室温で 2 時間。
- 65℃、10 分間で ligase を失活させる。
- 1-5 µl を形質転換 transformation に用いる。
古典的な ligation kit では、上記のように 10 - 20 µL の反応系を用いていたが、これについても効率化が進んでいる。たとえば Takara Ligation Kit Mighty Mix という試薬では、以下のように DNA 溶液とプレミックスを 1 : 1 で (ligation の種類によっては 1 + 2 で) 混ぜるだけである。
| ベクター + 挿入 DNA | 1 µL |
| Ligation Mix | 1 µL |
トラブルシューティング
コロニーが生えてこない場合に
制限酵素を使ったベクター構築をしている場合は、処理前の環状ベクターをポジティブコントロールに、処理後の直鎖状ベクターをネガティブコントロールにすることで、制限酵素処理が成功しているかどうかを確認することができる。もちろん、制限酵素処理産物の電気泳動 electrophoresis でも OK である。
問題点が ligation 反応にあるときは、以下の方法で改善する可能性がある。
DNA の損傷など
| 方法 | 文献 |
|---|---|
| 反応時間を延長する。 | 1 |
UV 照射によって DNA が損傷を受けるほか、ゲルからの DNA 抽出の際に残った不純物が ligation 反応を阻害する。エタノール沈殿によって状況が改善する場合がある。 |
1 |
| DNA が高次構造をとってしまっている可能性。突出末端ライゲーションの場合、ベクターとインサートの混合液を 60-65 ℃で数分加熱後、on ice で急冷してから buffer, ligase を加えることで効率が上がる場合がある。 | 1 |
DNA の精製が不十分なケース
| 方法 | 文献 |
|---|---|
| PCR 酵素の持ち込みによって、制限酵素反応中に断片が fill-in または deletion されている可能性。精製がエタノール沈殿のみでは、PCR 酵素が十分に取り除かれない。フェノール処理やカラム精製で改善するかもしれない。 | 2 |
その他のクローニング方法: TOPO, In-Fusion など
制限酵素と DNA リガーゼを使う方法は今や古典的な方法であり、インサートの大きさや組み替え効率を改善する方法が数多く存在する。
| TA cloning |
Taq が増幅末端に A を付加する性質を利用したクローニング。 |
| TOPO cloning |
DNA topoisomerase を使い、高い組み替え効率を実現。 |
| In-Fusion |
In-Fusion primer で増幅した PCR 産物を組み込む。DNA 断片末端の相同組み替えを利用しており、15 kB までの大きい断片の組み込みが容易である (6)。 |
広告
References
- Clontech, Takara Q & A. ライゲーション一般. Link.
- Bio Technical フォーラム. クローニングがうまくいきません. Link.
- Ligation protocol with T4 DNA ligase. NEB. Link
- タカラバイオ クローニング実験ハンドブック.
コメント欄
サーバー移転のため、コメント欄は一時閉鎖中です。サイドバーから「管理人への質問」へどうぞ。